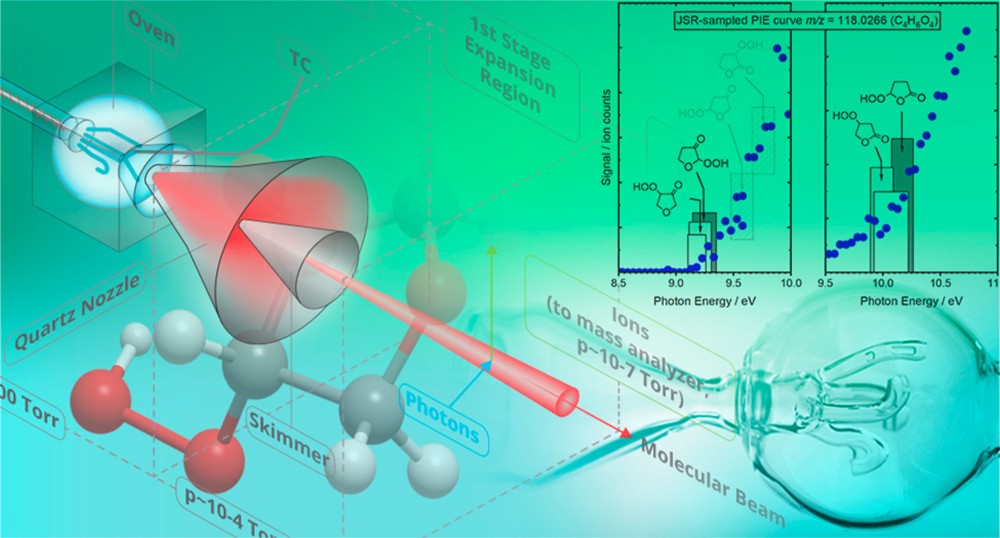
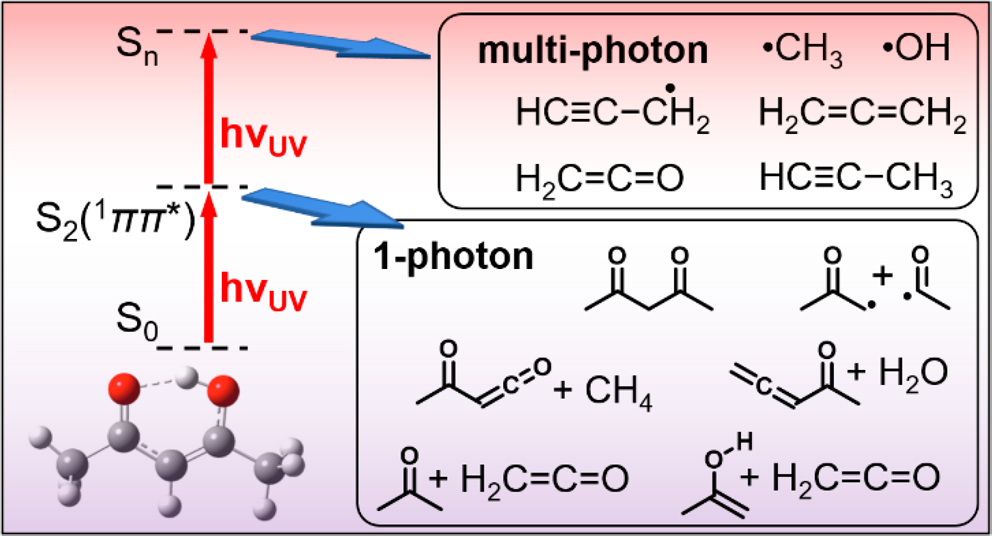
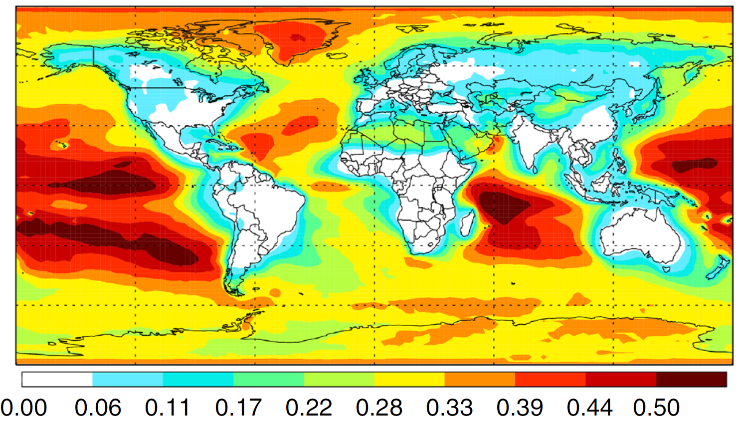
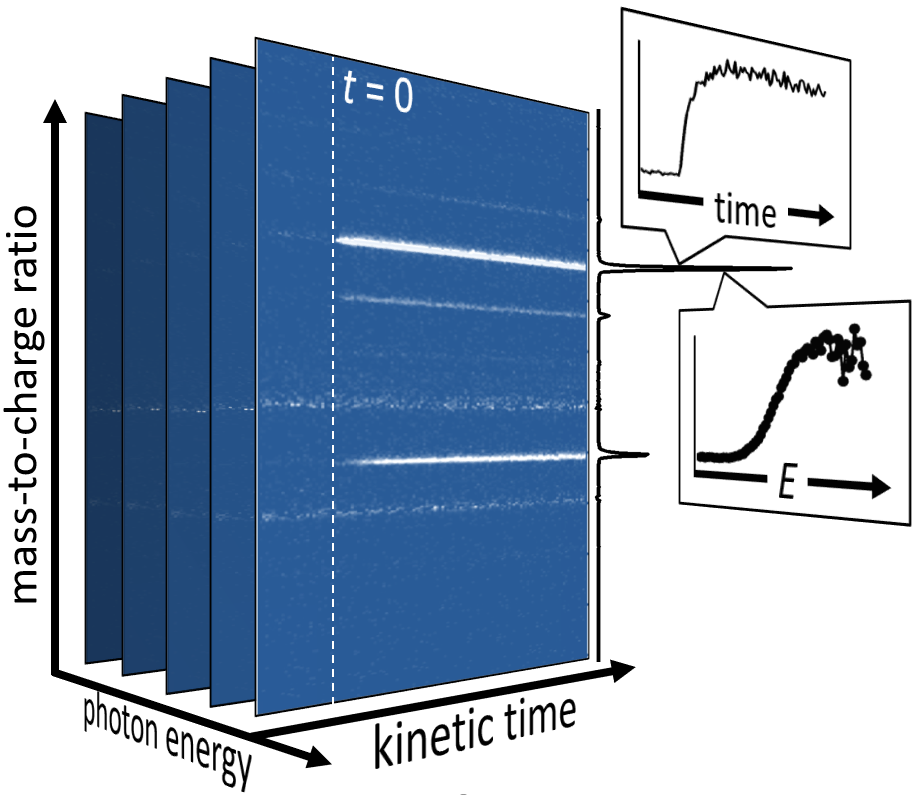
The CRF research into the fundamentals of chemical reactivity characterizes the building blocks important to all chemistry, thus contributing to a broad range of DOE energy missions. The work spans system complexity from dynamics experiments on single collision scattering or photodissociation of small molecules to detailed kinetics investigations of complex processes, e.g., autoxidation, described by a network of chemical reactions. The research employs multiplexed measurements to probe multiple observables simultaneously and is strengthened by close connections between experimental and theoretical investigations. Scientific themes include interactions on complex multi-well potential energy surfaces, reactants with unusual electronic character, characterization of elusive intermediates, reactions that couple multiple electronic surfaces, and effects of non-thermal reactant energy distributions.