CRF researchers have made breakthroughs that support chemical physics research in areas such as non-linear spectroscopy, ion imaging, 3-dimensional optical imaging, multiplexed photoionization/photoelectron spectroscopy, and x-ray absorption and scattering. Some of the resulting capabilities are described in the Experimental Capabilities section. Many of these breakthroughs are based in fundamental research on spectroscopy and light-matter interactions, which recently has concentrated on understanding non-linear effects. Intense pulsed laser fields impinging on atoms, molecules, and surfaces induce a coherent response in the material. At especially high intensities, the material response becomes nonlinear and gives rise to a wealth of approaches to control and probe molecular processes, even processes very far from chemical equilibrium. At the CRF, we develop ultrafast optical methods to measure and control matter at the molecular level and on molecular timescales. Intense lasers can spatially align molecules or coherently prepare highly excited molecular states. Ultrafast nonlinear spectroscopy is advanced for the detection of transient reactive molecular species, including broadband approaches that enable the sampling of many spectroscopic transitions at once and allow the assessment of instantaneous molecular energy partitioning and energy transfer.
Coherent Non-linear Spectroscopy
When strong optical fields interact with molecules, electron cloud polarization is induced. If a molecule’s polarizability is asymmetrical along different axes, the optical field will tend to align the molecules. If the pulse duration is longer than the rotational period of the molecules, adiabatic alignment will trap molecules in pendular states. By contrast, if the optical pulse is much shorter than the rotational period of the molecules, transient field-free alignment can be achieved. We use femtosecond/picosecond CARS signals as a sensitive probe to quantify the degree of alignment induced within the probed molecules.

Figure 1. One-dimensional fs/ps CARS image of adiabatic/nonadiabatic alignment effects. The y-axis is the vertical spatial coordinate along the laser sheet. The x-axis is the wavelength axis, and signal from J″=0 and J″=1 is labeled. Near the center of the sheet, overlap with the adiabatic alignment field is observed as a bright signal on the J″=1 line. A signal enhancement of as much as a factor of 120 was observed for the combined adiabatic/nonadiabatic alignment field.
The combination of adiabatic and nonadiabatic alignment fields has been studied in molecular H2, for example, and shown to exhibit a higher degree of molecular alignment than is achievable with either adiabatic or nonadiabatic alignment alone. Figure 1 displays a 1D fs/ps CARS image that is sensitive to the degree of molecular alignment. The vertical axis of this image corresponds to position along a line in space. The horizontal axis is the frequency of the signal. As the two populated rotational states of H2 are emitted at different frequencies, the signal from each rotational level is isolated in the two bright vertical signals observed. At the center of the probed region, a focused nanosecond nonadiabatic alignment field interacted with the molecules. A bright enhancement of the signal, signifying increased molecular alignment, is shown on the J”=1 rotational line within the image. This enhancement corresponds to the spatial location where the adiabatic aligning field was focused.
Key Contributions
- Coherent nonlinear optical spectroscopy is used both to control and probe the degree of molecular alignment.
- The combination of adiabatic and nonadiabatic molecular alignment approaches yields a higher degree of molecular control. We are presently implementing such approaches in the development of a refined optical centrifuge with increased control over the final rotational state distribution.
PIs: David L. Osborn, David W. Chandler, and Christopher J. Kliewer
Alignment of Molecules
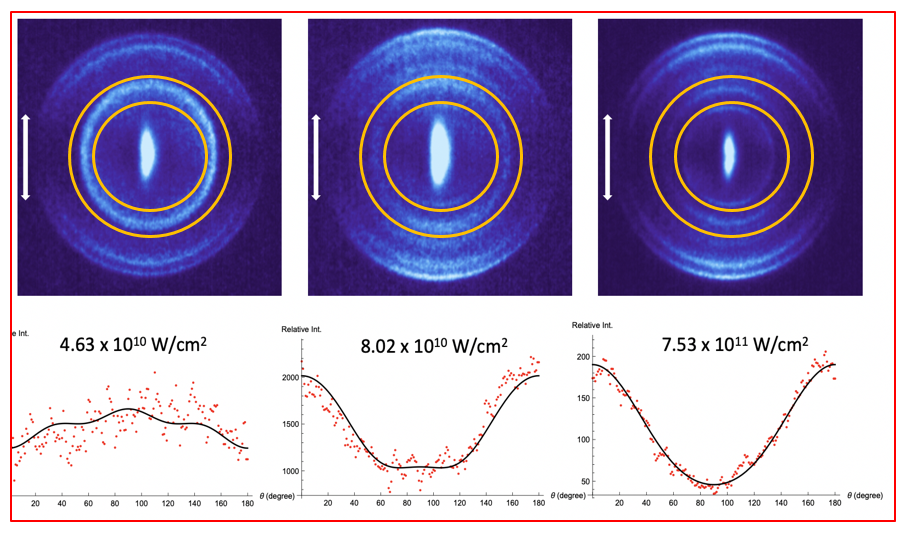
There are several accepted ways to align molecules using intense laser fields. If the laser is on for a long time compared to the rotational period of the molecule, adiabatic alignment is achieved where several molecular rotational states are mixed making a superposition state that is aligned. Adiabatic alignment has also been achieved utilizing static electric fields, sometimes called brute force alignment. The mechanism of interaction is through the polarization anisotropy of the molecule, which is the difference in the polarizability of the electrons along a molecular bond compared to perpendicular to the molecular bond. The molecule generally stays aligned as long as the laser field is applied. In non-adiabatic alignment a broadband laser pulse shorter than a rotational period excites a superposition of rotational states within an ensemble of molecules and within a single molecule, and as a function of times the superposition of states comes in and out of phase creating transient alignment at different times after the laser pulse. Both techniques have been widely used on the ground electronic states of molecules. We have demonstrated a new variant of adiabatic alignment where we utilize a mixture of excited valence states of a molecule. These states, with electrons in orbitals not as tightly bound to the nuclei as the ground electronic state electrons, show greater polarizability compared to ground electronic states, and we demonstrate that we are able to align molecular hydrogen at reduced laser intensity compared to that needed to align the hydrogen molecule in its ground electronic state. Rydberg states of molecules are known to have extremely large polarizabilities because the electrons are even more loosely bound to the molecule than excited valence states, but they are not useful for alignment because there is no polarization anisotropy associated with them (the molecule appears as a point charge within a large electron orbital).
After exciting the hydrogen molecule to the excited E,F electronic state, which has a lifetime of ~200 nanoseconds, we focus an 8-nanosecond long 1064-nm laser pulse onto the excited state molecules. This laser pulse mixes the E,F state with the nearby B and C states. Then, utilizing the polarization anisotropy of the resulting mixed state, the second laser aligns the H2 molecule to the laser polarization axis. By measuring the laser power dependence of the alignment we are able to determine the polarization anisotropy of the mixed state. This value is found to be (3.7 ± 1.2) x 103 a.u. This value is extremely large in comparison to what one would expect from either ground or the pure H2 (E,F) electronic state and an order of magnitude larger than the most polarizable ground state molecules that have been aligned. We speculate that this technique of utilizing excited valence states to achieve alignment could be widely applicable.