Technical Details
Ion imaging was first demonstrated at Sandia National Labs in 1987 by a collaborative research effort between Dr. David Chandler (Sandia) and Dr. Paul Houston (Cornell University). Since then, more than 100 other laboratories have adopted this technique for the study of photochemistry, bimolecular reactivity, energy transfer, dissociative electron attachment and electron scattering.
At Sandia, we have utilized this technique to study low energy secondary electrons emitted from an electron microscope. We have facilities to study unimolecular and bimolecular chemistry as well as electron driven processes (either electron scattering, dissociative electron attachment or laser driven plasmas).
We have also developed an electron imaging apparatus that allows one to study electron scattering under zero potential field and then quickly sweep the electron out of the region and image them to reveal inelastic electron scattering as well as study dissociative electron attachment processes. Once again, we have the capability to pre-excite the molecules with a laser before proceeding with the electron driven chemical process. This is a unique capability made possible by our use of laser produced electrons. The work is being supervised by David Chandler and Jonathan Frank.
Some of the “firsts” demonstrated in our laboratory include the first to demonstrate ion imaging (CH3I in 1987, collaboration with Paul Houston); the utilization polarized light to use imaging to measure the alignment of a product molecule as a function of angle for reaction products (CH3 from CH3I); the imaging of products of a bimolecular reaction (H + HI and H + D2); the measurement of the alignment and orientation of bimolecular products; and the measurement of the differential cross sections, alignment, and orientation of collision products of electronically excited state molecules.
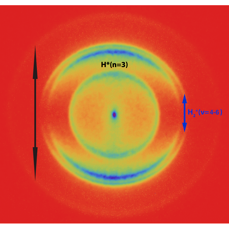
Figure 1 Image of protons ejected from H2 molecules after multiphoton excitation with intense UV (201 nm) and visible light (532 nm). The different rings indicate different channels of dissociation.
Electron and Ion Imaging for Study of Chemical Processes
The imaging of the charged products of unimolecular or bimolecular chemical processes (either collision or photon induced) provides deep understanding into the chemical dynamics of the process. Products can be charged as a natural consequence of the chemical process or laser ionized. The image below provides a measure of the velocity (speed and angle of motion) of the ion or electron and from this measurement the energetics, and often the mechanism, of the process can be determined.
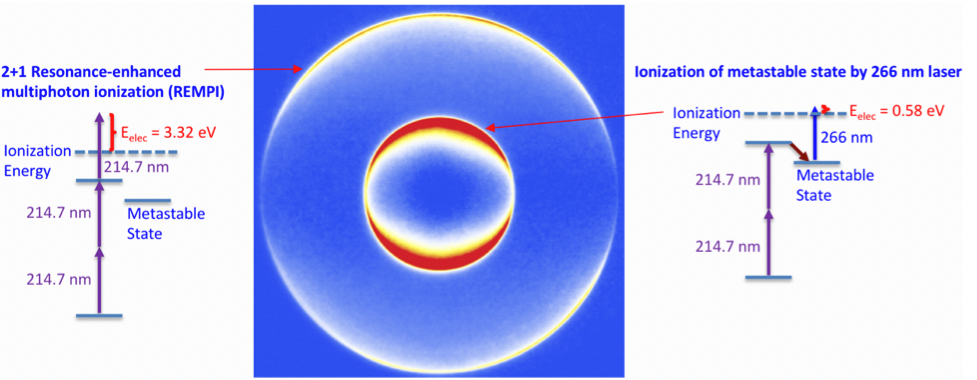
Figure 2 A simple example that demonstrates this power to determine the mechanism and energy of the process is shown in this image of electrons ejected by Kr atoms after multiphoton excitation. The Kr is excited with UV (214 nm) light and ultraviolet (266 nm) light and the two different rings are observed indicating two different processes that are shown next to the image.
The measurement of the velocity of a particular quantum state (rotational, vibrational and electronic state specified) of a chemical process gives unprecedented information about the process that produced that product. These studies, both at Sandia in the other laboratories) have provided information that has impacted combustion mechanisms, atmospheric photochemistry, the ozone hole formation, plasma processes, catalytic behavior at surfaces and the bonding of water and solvation mechanisms. These detailed studies provide data to rigorously test the best theories of chemical reactivity.
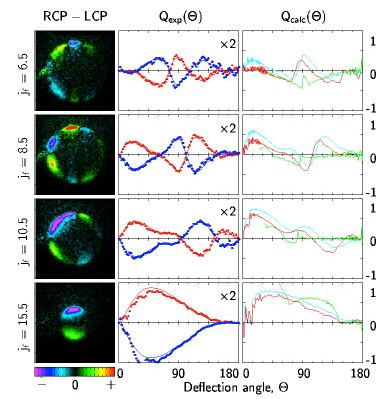
Figure 3 Images of specific quantum states of NO(jf) product after scattering NO(j=.5) from Argon and collecting the images with circularly polarized light. The patterns indicate that the product molecules in specific quantum states scattering at specific angles have a propensity to rotate either clockwise or counterclockwise and this propensity alternates more quickly with low rotational state products than with high rotational state products.
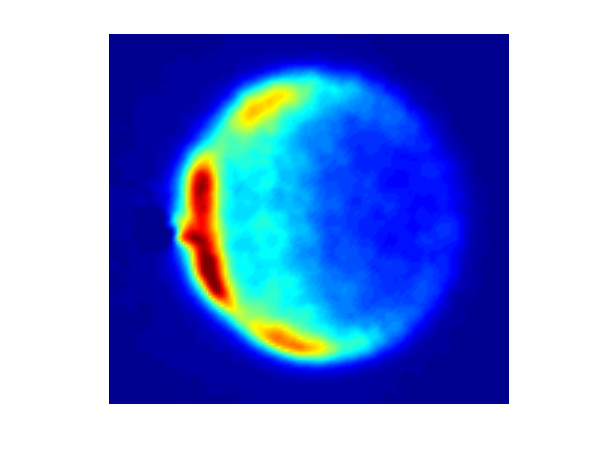
Figure 4 Image of NO(A, j=7) scattering r NO(A, j=0) is scattered from Ar atoms. In these experiments the NO is excited to the A electronic state that lives for 200 ns and during that time the NO(A) has a collision and a single quantum state of the product of that collisional excitation is ionized and detected, in this case the j=7 rotational state of the A electronic state.
Partners
A partial list of recent collaborators includes Dr. Richard Zare, Stanford; Dr. Peter Rakitzis, University of Crete; Drs. Mathew Costen and Kenneth McKendrick, Harriot Watt University; and Dr. Javier Aoiz, Madrid