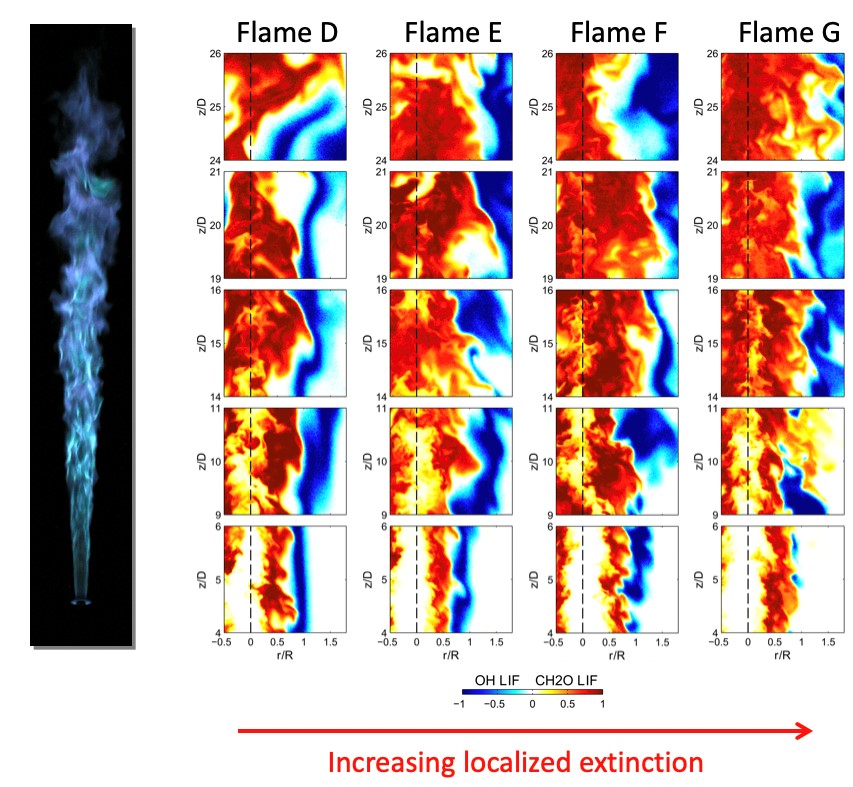
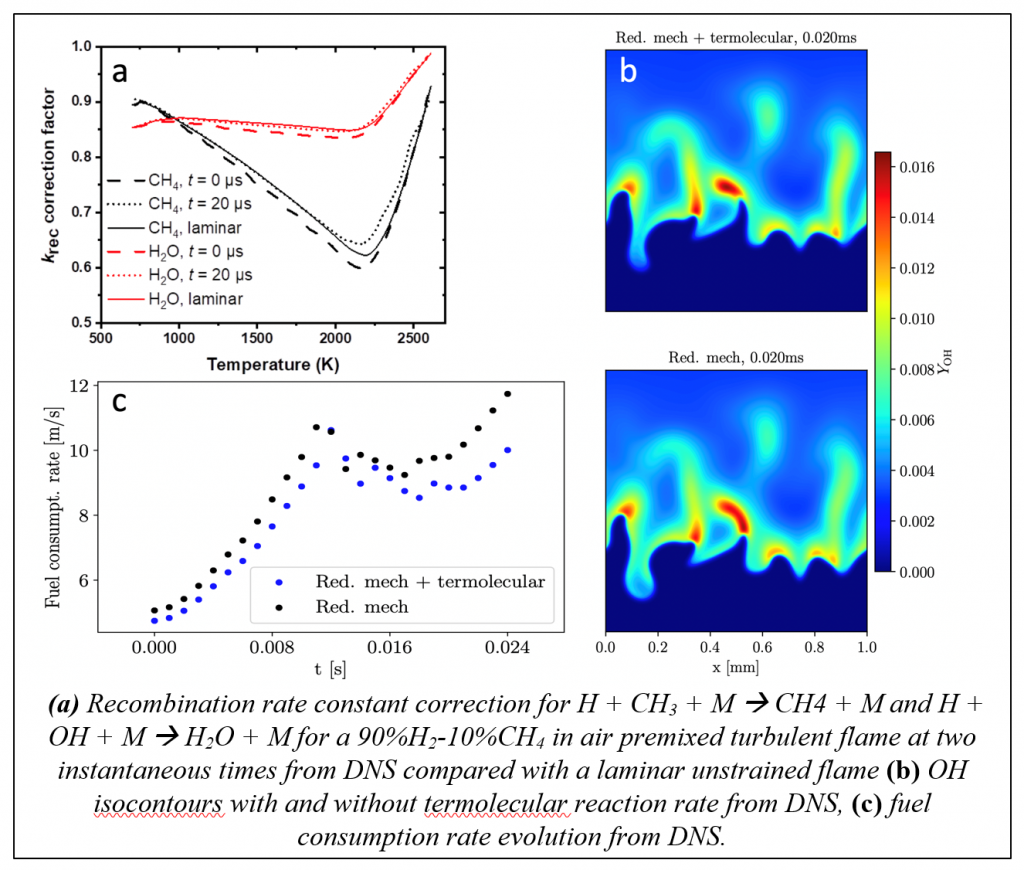
Combustion systems are characterized by a complex interplay between convection and diffusional transport processes and chemical reaction rates, particularly in turbulent flows, wherein strong, time-varying gradients in both chemical composition and temperature couple with chemical reactions. These systems are practically relevant and scientifically challenging to investigate and understand, due to their wide range of relevant spatial and temporal scales. The CRF program has provided decades of leadership in combining experiments and modeling of turbulent combustion through advanced diagnostics development, leading-edge modeling and simulation, and collaborations within the greater combustion research community, as exemplified by the International Workshop on Measurement and Combustion of Turbulent Flames. Current work in the interactions of chemistry and transport focuses on connecting continuum flow to the molecular level, building from detailed studies of elementary non-reactive collisions to modeling the connection of spatial temperature gradients to non-equilibrium chemistry.