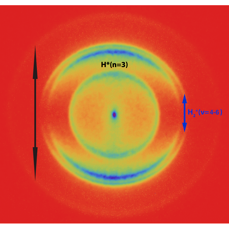
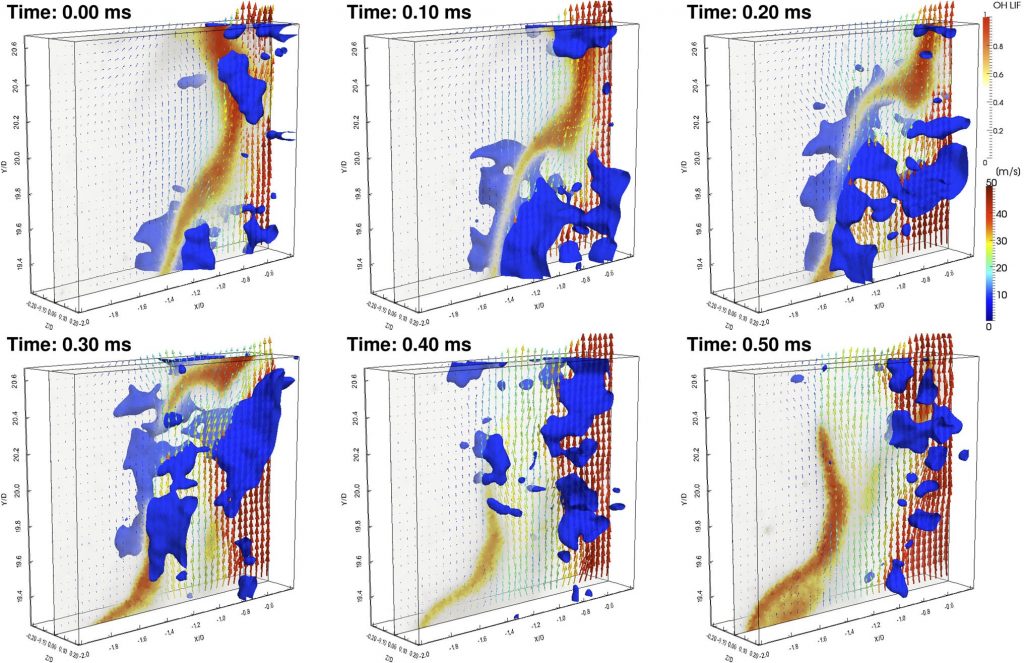
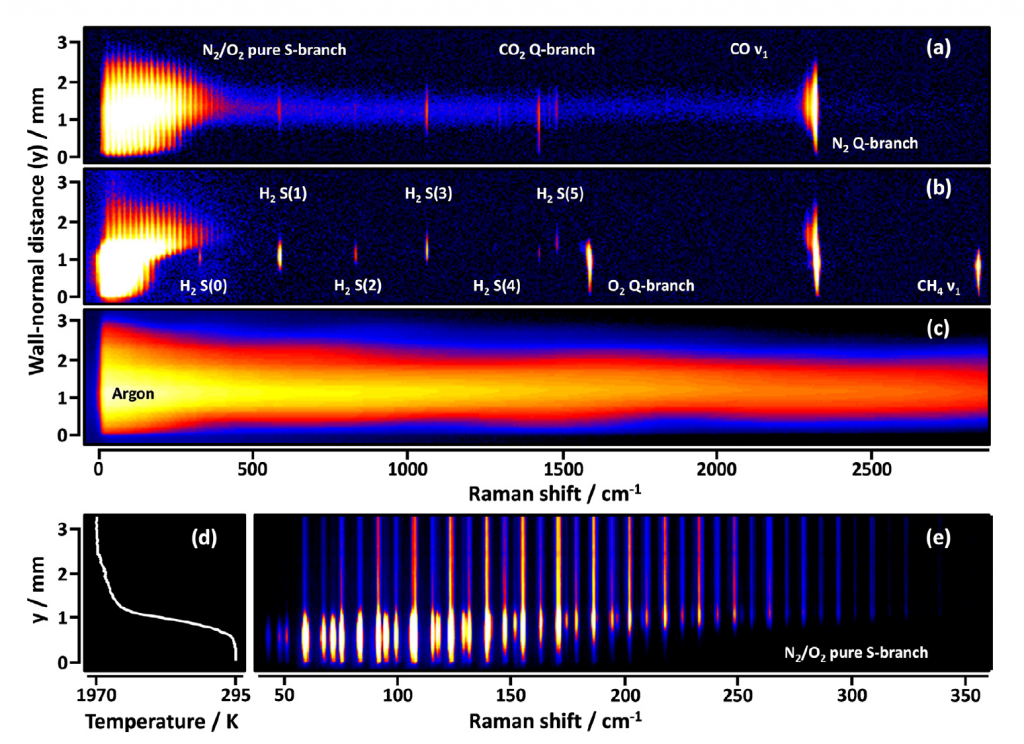
Understanding complex chemical physics systems often involves correlations among properties, making multidimensional diagnostics a natural tool. The CRF invents and employs imaging methods to probe such correlations: from ion imaging, which directly measures correlated angular and velocity distributions in single molecular collisions or photodissociation to four-dimensional imaging of chemical processes in turbulent flames.